4.3 Sex specific genetic variance and inter-sexual genetic correlations
ds <- data.frame(animal=gryphons[,"id"],sex=sample(c("Female","Male"),nrow(gryphons), replace=TRUE))
squid_data <- simulate_population(
parameters = list(
sex=list(
fixed=TRUE,
names=c("Female","Male"),
beta=c(-0.5,0.5)
),
animal= list(
names = c("G_female","G_male"),
vcov =matrix(c(0.1,-0.1,-0.1,0.4), nrow=2, ncol=2 ,byrow=TRUE)
),
residual = list(
names="residual",
vcov = 0.1
)
),
data_structure = ds,
pedigree = list(animal=ped),
model = "y = Female + Male + I(Female)*G_female + I(Male)*G_male + residual"
)
data <- get_population_data(squid_data)
head(data)## y Female Male G_female G_male residual animal sex
## 1 -1.1406269 1 0 -0.41650798 0.8611695 -0.22411895 204 Female
## 2 -0.9338681 1 0 0.02895999 0.7161771 -0.46282805 205 Female
## 3 -0.5726704 1 0 -0.11908647 0.3606215 0.04641609 206 Female
## 4 0.5511020 0 1 -0.45076138 0.2253053 -0.17420333 207 Male
## 5 -0.6838055 1 0 -0.12133713 -0.3310633 -0.06246839 208 Female
## 6 -0.1827386 0 1 0.33321284 -0.6581778 -0.02456084 209 Male
## squid_pop
## 1 1
## 2 1
## 3 1
## 4 1
## 5 1
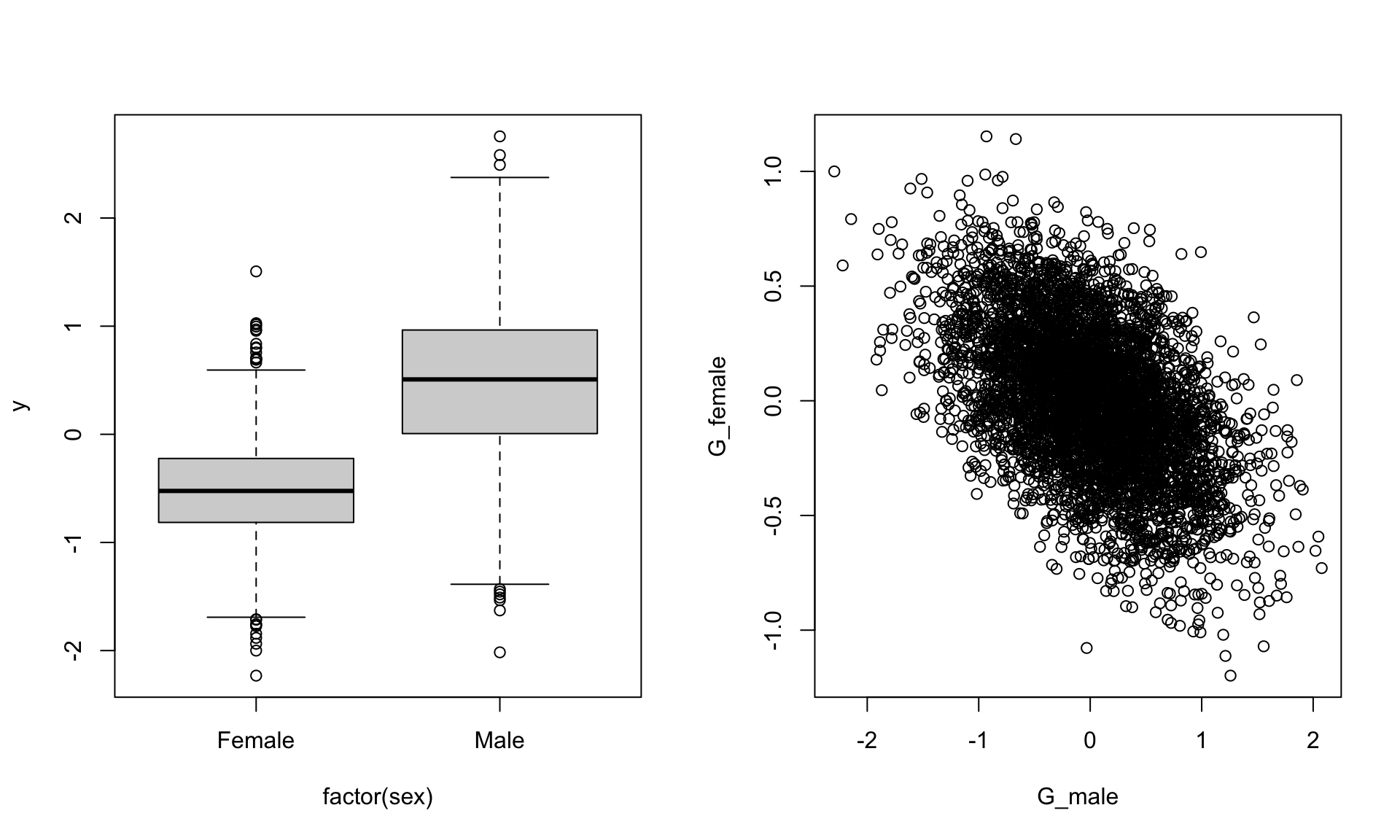
## 6 1par(mfrow=c(1,2))
boxplot(y~factor(sex),data)
plot(G_female~G_male,data)